Trim adapters from reads in fastq file
In order for DNA to be read by the sequencer, adapters are added to each end of the DNA sequence. Most modern sequencers recognise the adapters and remove these automatically but older sequencers do not, so the adapters need to be removed manually. If adapters are left in place your read alignment percentages will be very low when you come to align the reads your fastq files.
I have only done this once so far, this was on paired end data and I used the package Trim Galore! to do it.
mkdir -p trimmed_fastq_files
trim_galore --nextera -q20 --paired -o trimmed_fastq_files --fastqc --gzip sample1_R1.fastq.gz sample1_R2.fastq.gz
As we didn’t create an output folder to store trimmed fastq files initially make sure to create a new directory first, then run trim galore on all the files. Trim galore automatically creates a fastqc report on any file it attempts to trim adapters from (by adding the –fastqc flag).
This means all we have to do is run multiqc again on the new trimmed fastqc html files.
# Either from the home directory
multiqc trimmed_fastq_files/
# OR
cd trimmed_fastq_files
multiqc .
As before visualise the html on a web browser.

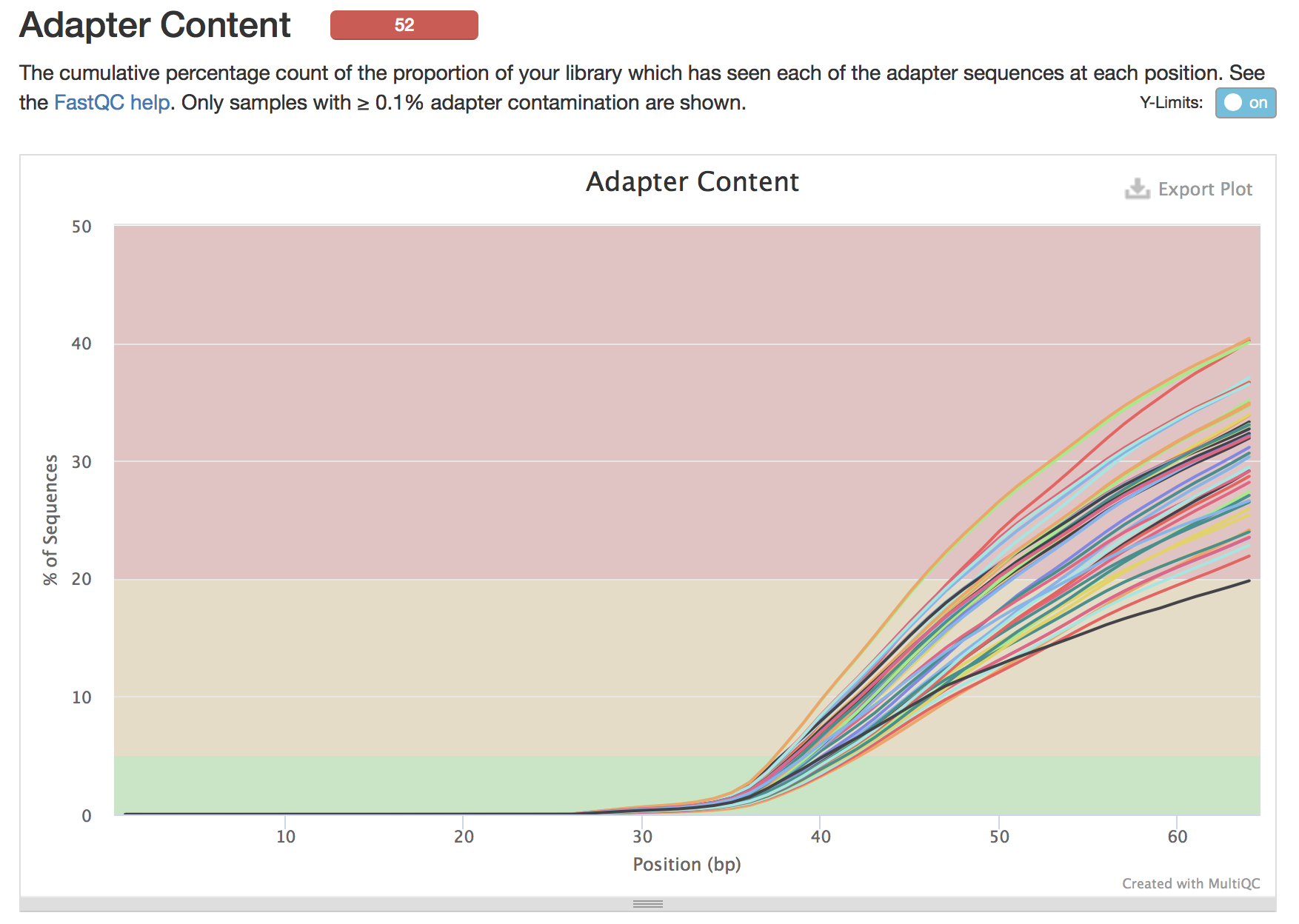
You can see that the adpaters have been successfully removed.
Move on to Alignment, or back to Quality control - FastQC/MultiQC.