Collecting SRA file information from a public repository
What is an SRA file?
SRA stands for sequence read archive. SRA files are stored on bioinformatics databases that store DNA
sequencing data. SRA files are stored in many formats .bam, .bed etc. but, in this tutorial,
we are interested in downloading .fastq SRA files.
Where are the files stored?
The Gene Expression Omnibus (GEO) is a public functional genomics data repository containing array- and sequenced based data. Although other repositories exist, some which require access permissions, many groups opt to post their data on GEO.
Obtaining SRA files from GEO repositories can be quite confusing. See here for tips. The best method I’ve found so far to download files is described below.
We need to find the SRA numbers for the files we are interested in and annotate them in a sensible way so we can keep track of the files as they move through the pipeline and to easily recognise them if we come back to look at them 2 years from now. This is probably the most important part of the whole process as you can easily make a typo somewhere and download a completely different file from a different study!
Open a text editor
We start by opening a text file in our home folder.
nano sra_files.txt
This opens a new .txt file called ‘sra_files’ in the text editor called nano. Nano has basic functionality,
many use vim, but nano is fine for our purposes here. We want to copy all the SRA numbers of the SRA files
we’re interested in into this file.
Leave the nano window open, and in your web browser navigate to this GEO page in a separate tab (use the GEO
link above).
Obtain SRA files from GEO repository for study of interest
Normally you’ll be directed to a GEO repository from a paper you are interested in. This will provide you with a GEO accession number like GSEXXXXX which is the code for the central page containing the data associated with the study.
The accession number for our study is GSE74912. Type this into the box that says ‘Keyword or GEO Accession’ and hit ‘SEARCH’.
This takes you to the main page for our study of interest. This page provides a brief summary of the study, contact details for the PIs and, if you scroll down to the ‘Samples’ section and hit ‘More’, you can see what types of data are stored in the repo. We can see there are data for 31 samples in total with 130 ATAC-seq samples.
Go back to the top of the page and click the Query DataSets for GSE74912 link.
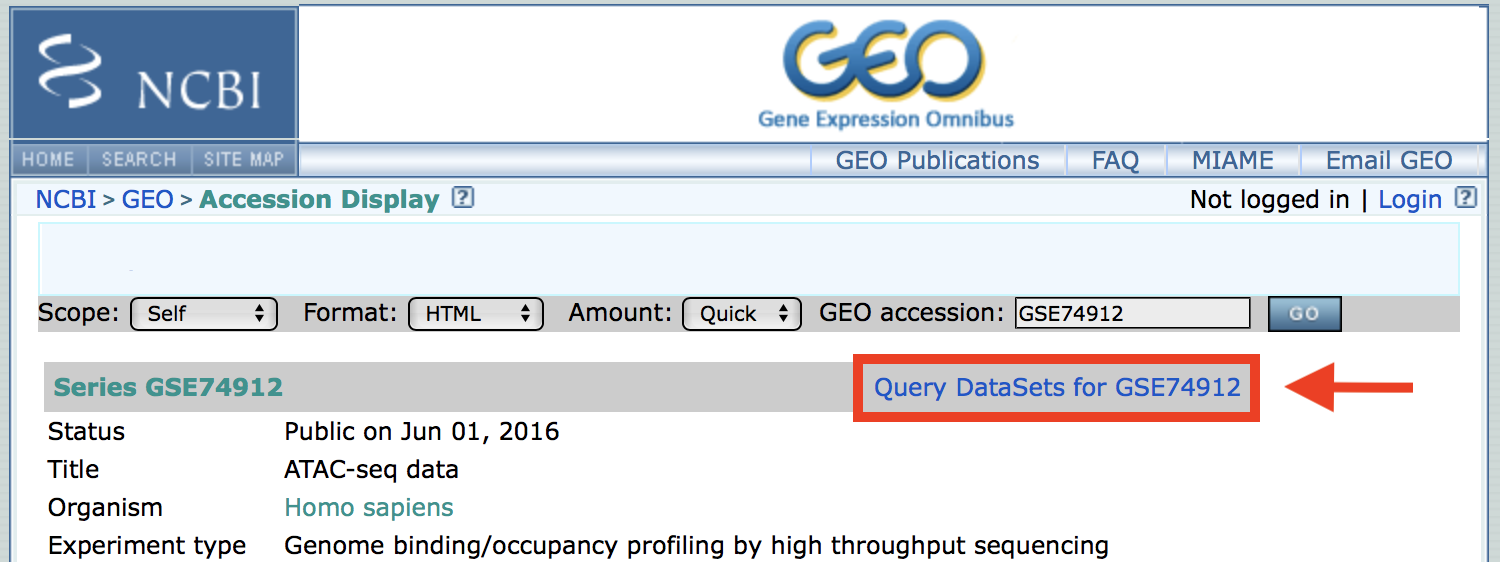
This takes you to another list of all the replicates in the repo. We want the SRA numbers for files 10-26 inclusive.
Navigate to file 10, which is the first ChIP-seq dataset, and select the ‘SRA Run Selector’ link.
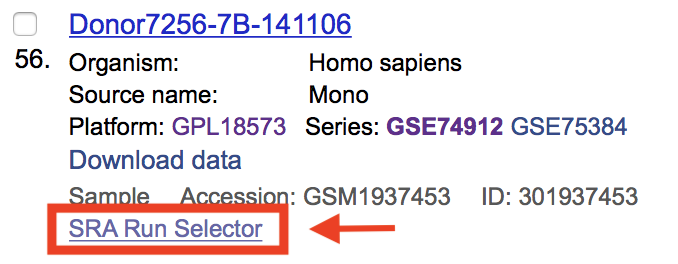
This takes us to the page containing the SRA file number/s for that replicate. The SRA number we need is listed next to Run: and is
SRR1647895.
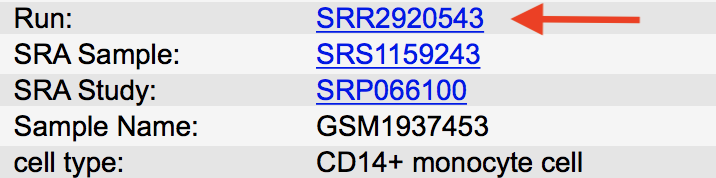
Note: Occasionally there are more than one SRA files per replicate, this means that the data was generated over multiple lanes in the sequencer and the files would need to be merged in the in the pipeline.
There is some other important information here. Notice under LibraryLayout: it says PAIRED this means the data
is paired ended rather than single ended, we need to know this as it will influence some of the parameters we need to
specify later on.
For now copy and paste the SRA numbers for 4 replicates into the text file one file per line.
SRR2920543
SRR2920542
SRR2920521
SRR2920520
The first two SRA numbers refer to CD14+ Monocyte ATAC-seq data files and the last two to CD8+ T cell ATAC-seq data files. These data were derived from cells extracted from peripheral blood and are entries 56,57, 78 and 79 in the list of files.
As alluded to earlier, it is important to keep an accurate record of the replicate information each SRA number refers to. This study is annotated fairly clearly, but this is not always the case. At this stage I usually create a table containing all the replicate information using abbriviated IDs. These IDs are substitued into the file names instead of the SRA numbers to make it easier to see what files are affected during the pipeline.
To save and close the text file in nano press:
ctrl x
y
then enter
Move on to Download SRA files, or back to Getting started.